
This past month has been a banner month for Machine Learning as three key reports have come out that change the way that the average lay person should think about machine learning. Two of these papers are about conducting machine learning while considering underspecification and using deep evidential regression to estimate uncertainty. The third report is about a stunning result in machine learning’s role to improve protein folding.
The first report was written by a team of 40 Google researchers, titled Underspecification Presents Challenges for Credibility in Modern Machine Learning. Behind the title is the basic problem that certain predictors can lead to nearly identical results in a testing environment, but provide vastly different results in a production environment. It can be easy to simply train a model or to optimize a model to provide a strong initial fit. However, savvy machine learning analysts and developers will realize that their models need to be aligned not only to good results, but to the full context of the environment, language, risk profile, and other aspects of the problem in question.
The paper suggests conducting additional real-world stress tests for models that may seem similar and to understand the full scope of requirements associated with the model in question. As with much of the data world, the key for avoiding underspecification seems to come back to strong due diligence and robust testing rather than simply trusting the numbers.
The second report is Deep Evidential Regression, written by a team of MIT and Harvard authors which did the following.
In this paper, we propose a novel method for training non-Bayesian NNs to estimate a continuous target as well as its associated evidence in order to learn both aleatoric and epistemic uncertainty. We accomplish this by placing evidential priors over the original Gaussian likelihood function and training the NN to infer the hyperparameters of the evidential distribution
http://www.mit.edu/~amini/pubs/pdf/deep-evidential-regression.pdf
From a practical perspective, this method provides a relatively simple way to understand how “uncertain” your neural net is compared to the reality that it is trying to reflect. This paper moves beyond the standard measures of variance and accuracy to start trying to understand how confident we can be in the models being created. From my perspective, this concept couples well with the problem of underspecification. Together, I believe these two papers will help data scientists go a long way towards cleaning up models that look superficially good, but fail to reflect real world results.
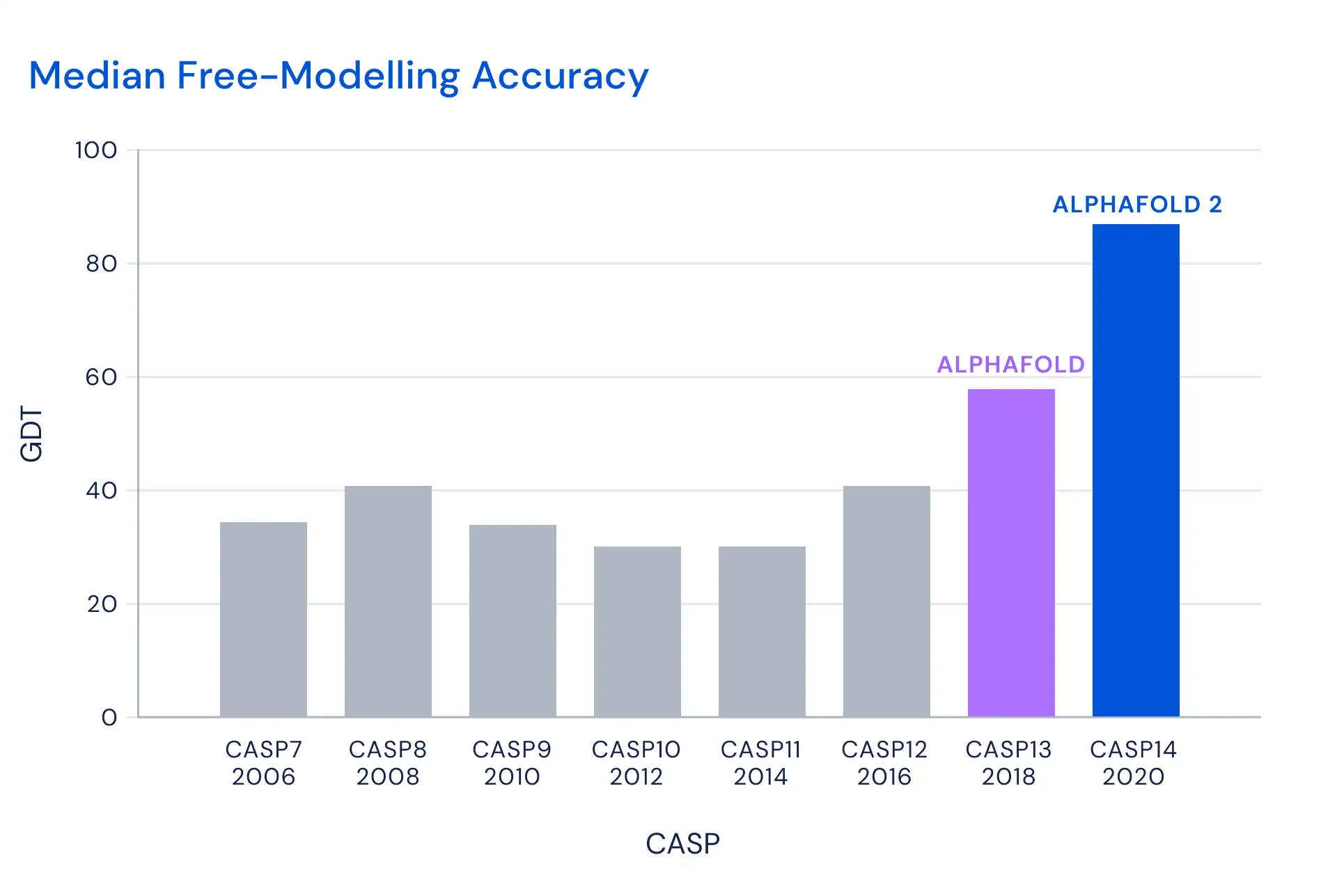
Finally, I would be remiss if I didn’t mention the success of DeepMind’s program, AlphaFold, in the Critical Assessment of Structure Prediction challenge, which focuses on protein-structure predictions.
Although DeepMind has been working on AlphaFold for years, this current version tested yesterday provided results that were a quantum leap compared to prior years.
The reason that protein folding is so difficult to calculate is that there are multiple levels of structure to a protein. We learn about amino acids, which are the building blocks of proteins and basically defined by DNA. The A’s, T’s, C’s, and G’s basically provide an alphabet that defines the linear lineup of a protein with groups of three nucleotides defining an amino acid.
But then there’s a secondary structure where internal bonding can make the proteins line up as alpha sheets or beta helices. The totality of these secondary structures, this combination of sheets and helix shapes, makes up the tertiary structure.
And then multiple chains of tertiary structure can come together into a quaternary structure, which is the end game for building a protein. If you really want to learn the details, Khan Academy has a nice video to walk you through the details, as I’ve skipped all of the chemistry.
But the big takeaway: there are four levels of increasingly complicated chemical structure for a protein, each with its own set of interactions that make it very computationally challenging to guess what a protein would look like based just on having the basic DNA sequence or the related amino acid sequence.
Billions of computing hours have been spent on trying to figure out some vague idea of what a protein might look like and billions of lab hours have then been spent trying to test whether this wild guess is accurate or, more likely, not. This is why it is an amazing game-changer to see that DeepMind has basically nailed what the quaternary structure looks like.
This version of AlphaFold is an exciting Nobel Prize-caliber discovery. I think this will be the first Nobel Prize driven by deep learning and this discovery is an exciting validation of the value of AI at a practical level. At this point, AlphaFold is the “Data Prep” tool for protein folding with the same potential to greatly reduce the effort needed to simply make sure that a protein is feasable.
This discovery will improve our ability to create drugs, explore biological systems, and fundamentally understand how mutations affect proteins on a universal scale.
This is an exciting time to be a part of the AI community and to see advances being made literally on a weekly basis. As an analyst in this space, I look forward to seeing how these, and other discoveries, filter down to tools that we are able to use for business and at home.